
Perween Mufti ( Department of Paediatrics, Aga Khan University Hospital, Karachi. )
Iqbal Ahmed ( Department of Paediatrics, Aga Khan University Hospital, Karachi. )
November 1993, Volume 43, Issue 11
Original Article
Abstract
A total of 21 patients were admitted to Aga Khan University Hospital with suspected congenital hyperammonemias during the period 1989 to 1992, 11 with acidosis and 10 without acidosis. Prominent clinical manifestations included positive family history (76%), onset in the first week of life (67%) and neurological manifestations (76%). Of patients with hyperammonemia and acidosis, 4 had severe metabolic acidosis with anion gap of 3OmEq/L and above. Of patients with hyperammonemia without acidosis, 4 had ammonia level ranging from 1600-2000ug/dl. Diagnosis was confirmed in only 1 patient and that was also done abroad. Overall mortality was 71%. These disorders are not uncommon in our country and should be suspected in all Infants with above clinical or biochemical abnormalities (JPMA 43:232, 1993).
Introduction
Inborn errors of metabolism are a group of disorders which result from partial or complete absence of enzymes involved in biochemical reactions within the cells. This leads to both abnormal synthesis as well as ketabolism of metabolites. Most of these metabolites are neurotoxic and may cause death in early neonatal period or severe neurological disability. There are about 60 inherited metabolic disorders which can present in the neonatal period1,2. Number of these can be treated successfully if suspected and diagnosed early. Galactosemia3, phenylketonuria4, homocystinuria5,6, methylmalonic aciduria6,7 and congenital hyperammonemia8-10 are group of disorders for which treatment is available. This study reports different types of disorders with hyperammonemia, their clinical manifestation and outcome.
Methodology
From January, 1989 to December, 1992, a review of charts of all infants whose laboratory evaluation or clinical manifestation were suggestive of congenital hyper ammonemias were included in the study. Clinical manifestations and laboratory evaluation, which helped in the diagnosis are shown in Tables I and II1,2,11-13.

Results
Twenty one (11 with and 10 without metabolic acidosis) patients, suspected of having hyperammonemia (Table III).
Patients with isovaleric acidemia and nonketotic hyperglycinemia diagnosed elsewhere were followed at AKU. Ml infants were admitted to neonatal intensive care unit/intensive care unit.
Table IV shows the treatment given to these infants.8,10,12,14,15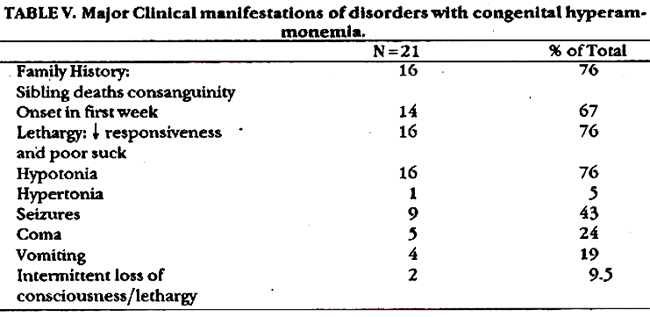
Table V shows major clinical manifestation. Prominent fntures included positive family history (76%), onset n the first week of life (67%) and neurological manifestation (76%).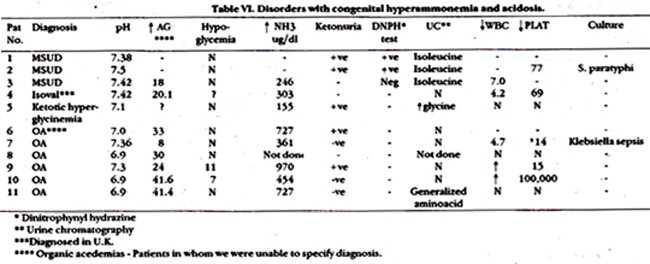
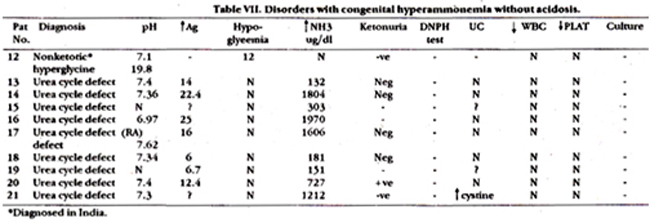
Table VI and VII show laboratory evaluations of these patients. All 3 patients suspected of having Maple syrup urine disease (MSUD) had abnormal excretion of isoleucine. 1/3 infants also had Maple syrup body odour. Ammonia level was normal in 2/3 infants. Of 6 patients suspected of organic acidemia, five died. Four had severe metabolic acidosis with aniongap of> 3OmEaJl. Patient # 7 was included in the study because there was strong family history of sibling deaths, compensated metabolic acidosis and high sepsis. This infant is alive and thriving. Urinary chromatography was not helpful in any of these cases. Among infants with hyperammonemia without acidosis, 4 had ammonia level ranging from 1600-nearly 2000 ug/dl. One of these infants also had terminal metabolic acidosis with pH of 6.9 and an aniongap of 25 mEq/I (Patient # 14,16,17,21). Patient # 21 was first seen at 8 months of age with recurrent episodes of intermittent vomiting and drowsiness and then at one year of age. This time his drowsiness progressed to coma. Ammonia level prior to death was 727 ug/dl. Urea cycle defect was suspected in all 5 infants. The other 4 infants were also suspected of having urea cycle defect. Patient # 13 was twin brother of patient # 16; he died at 2 months of age in a comatosed state. Unfortunately NH3 level was not documented at this time. Patient # 18 presented with failure to thrive. vomiting and diarrhoea. He weighed 1.9 kg at 2 months of age. This child was lost to follow-up. Patient # 19 developed seizures at I month of age. Three of her siblings died of similar complaints. Patient # 14 was aged 4 years who had intermittent episodes of unconsciousness, was lost to follow-up. Infant with non-ketotic hyperglycinemia was 2 year old female who presented to ER with hypoglycemic coma, and died on the same day.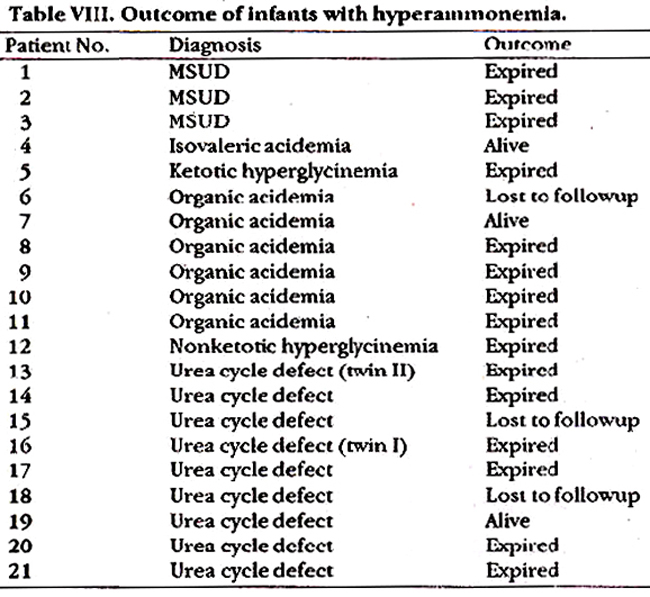
Table VIII shows the outcome. 15/21 (7 1%) infants died. Three infants were lost to follow-up.
Discussion
This is undoubtedly large group of patients with congenital hyperammonemia seen at AKU which is one of the largest teaching hospitals in this country. One of the major problems encountered here was confirmation of diagnosis which could not be done because of non existence of diagnostic facilities needed for this purpose. However, inspite of this, these conditions were suspected on clinical symptom and tests described in Table II. Overall mortality was extremely high. In patients with less severe clinical presentation proper management led to improvement. It is felt that because of consanguineous marriages in this country these problems may not be infrequent and there is a great need for establishing diagnostic facilities to confirm diagnosis, institute early treatment for better outcome and counselling parents. It is important to make the diagnosis for the sake of parents who have every right to know why their infants had died and for the purpose of genetic counselling.
References
1. Burton, B.K. and Nadler, H.L. Clinical diagnosis of the inborn errors of metabolism in the neonatal period. Pediatrics, 1978;61:398-405.
2. Burton, BK. Inborn errors of metabolism: the clinical diagnosis in early infancy. Pediatrics, 1987;79:359-69.
3. Donnell, G.N., Koch, R. and Bergen, Wit Observations on results of management of galactosemic patients in Hsia DYY (ed). Galactosemis Springfield, Charles C. Thomas Publisher, 1969; pp. 247-75.
4. Clayton, B.E., Moncrieff, A. and Roberts, G.E. Dietetic treatment of phenylketonuria: a follow-up study. Br.Med.J., 1967;3:133-36
5. Gaull, G.E. and Sturman, J.A., Vitamin B12 dependent methylmalonicaciduria: amino acid toxicity, long chain ketonuria and protective effect of vitamin B12 Br. Med.J., 1971;3:532-33.
6. Rosenberg, L.E. Inherited aminoacidopathies demonstrating vitamin dependency. N.Engl.J.Med., 1969;281:145-53.
7. Hsia, YE, Lilljeqvist, A-Ch and Rosenberg, Lit Vitamin B12 dependent methylmalonicaciduria. Pediatrics, 1970;46:497-507.
8. Batshaw, M.L, Brusilow, S., Wsber, L et al. Treatment of inborn errors of urea synthesis. N.Engl.J.Med., 1982;306:1387-92.
9. Batahaw, M.L and Brusilow, SW. Treatment of hyperammonemic coma caused by inborn errors of urea synthesis. J. Pedistr., 1980;97:893-900.
10. Bruaillow, S.W., Danney, M., Weber, Li. et al. Treatment of episodic hyperammonemia in cbildrenwith inborn errors of urea synthesis. N.EnglJ.Med., 1984;310:1630-34.
11. Green, A. and Hall, S.M. Investigation of metabolic disorders resembling Reye\\\'s syndrome. Arch. Dis.Child., 1992;67:1313-17.
12. Wraith, J.E. Diagnosis and management of inborn errors of metabolism. Arch. Dis. Child., 1989;64:1410-15.
13. Kronick, J.B., Scriver, C.R., Goodyer, P.lt et al. A perimortem protocol for suspected genetic disease. Pediatrics, 1983;71(6):960-63.
14. Dixon, M.A. and Leonard, J.V. Intercurrent illness in inborn errors of intermediary metabolism. Arch. Dis. Child., 1992;67:1387-91.
15. Naglak, M., Salvo, It, Madsen, K et al The treatmentof isovaleric acidemia with glycine supplement. Pedistr. Rca., 1958;24(1):9-13.
Journal of the Pakistan Medical Association has agreed to receive and publish manuscripts in accordance with the principles of the following committees: