
Fazal Manzoor Arain ( Department of Biological and Biomedical Sciences, The Aga Khan University, Karachi. )
Prem Chand ( Department of Pediatric Neurology, The Aga Khan University, Karachi. )
October 2015, Volume 65, Issue 10
Case Reports
Abstract
Hereditary Sensory Autonomic Neuropathy II (HSAN II) is a rare genetic disorder, characterized by severe loss of pain, temperature and touch sensation. Injuries in these patients can progress to necrosis and shedding of digits and limbs. Here we report two cases of HSAN II belonging to a Pakistani family. Individual 1, a forty five year old man, had complete loss of pain sensation since birth. Self-mutilation and complication of injuries resulted in the shedding of all the digits and right foot and surgical amputation of left leg. Individual 2, a five year old girl,had delay in healing of wounds and self-mutilation. Examination showed a complete lack of pain sensation throughout her body and hyporeflexia. As the genetic cause of HSAN II is unknown, identification of more patients will allow further research on this disease and possibly develop a cure.
Keywords: Genetic, Neuropathy, Pain, Self-mutilation.
Introduction
Hereditary Sensory Autonomic Neuropathy (HSAN) type I-V is a group of five clinically heterogeneous disorders whose phenotype ranges from purely sensory deficit to variable levels of motor and autonomic disturbances.1 HSAN are rare genetic disorders that show autosomal recessive mode of inheritance, except HSAN I which is autosomal dominant.2 The genes for all of these disorders have been discovered, except for HSAN II which remains unknown. A few candidate genes for HSAN II have been identified, but none could be associated to every case of HSAN II. That is why the diagnosis of HSAN II is still only based on clinical presentation.3 Patients diagnosed with HSAN II have a severe loss of pain, temperature and touch sensation since birth.4 Loss of these sensations can leave fractures of bones and even minor injuries unnoticed until they develop severe infections and might lead to amputation of digits or even limbs. Self-mutilation may begin soon after eruption of primary dentition.3 In some patients reduced tendon reflexes and mild autonomic disturbances that include swallowing problems, lack of tearing and slow pupillary response to light, may also be observed.1,5 Interestingly the remaining neurological exam, including mental functioning, muscle strength and power, is normal. Hence the neurological abnormality in these patients is restricted to sensory and autonomic nervous system.
Mutations in few candidate genes, including WNK1, K1F1 and FAM134B, have been associated with HSAN II.1 However the role of these mutations in the manifestation of this disease is not known. More cases of this disease need to be identified so that better genetic studies can be performed to identify the precise gene involved in this disease and how its dysfunction can result in the manifestation of this disorder. Here we report the findings from two individuals diagnosed of HSAN II who belong to a large Pakistani family.
Case Report
Two cases of HSAN II presented in September 2014. Patient I was a forty five year old male with a long standing diagnosis of HSAN II. Since childhood the patient did not have any pain sensation, which resulted is his minor and major injuries to go unnoticed for long periods. Severe infections, bone fractures and self-mutilation eventually led to shedding of digits of both hands (Figure-1 A and B) and the distal end of right foot (Figure-1 C). Due to necrosis and infection the left foot had to be surgically amputated above the ankle (Figure-1 D).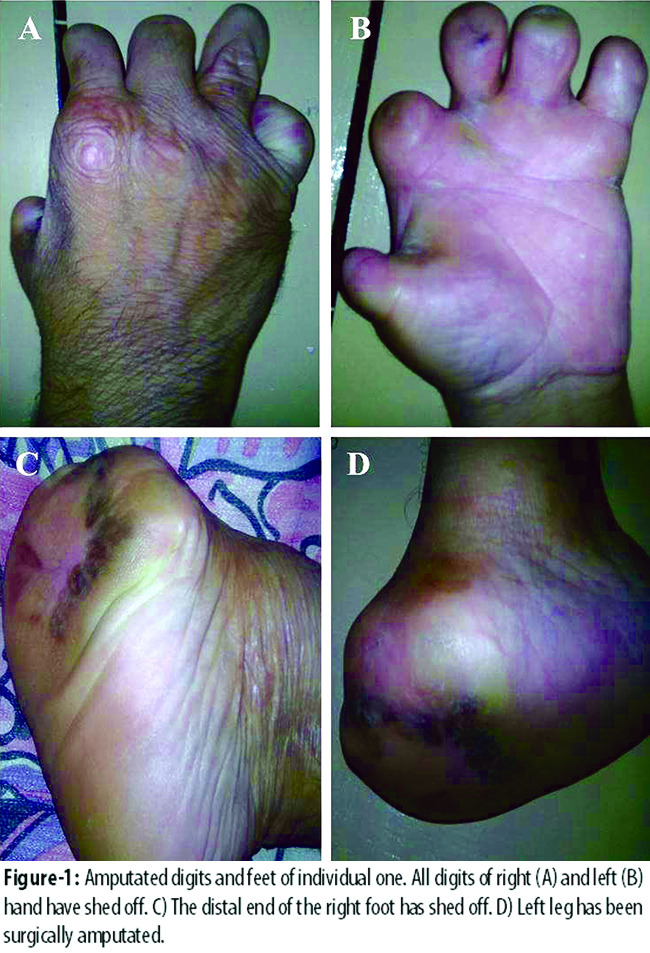
These events left the individual severely handicapped. However, no other systematic or neurological deficit was reported. Younger brother of the patient was also diagnosed of HSAN II and also had shedding of digits and limbs. The brother died at the age of 14 due to unknown cause.
Patient I was also the maternal uncle of patient II (the pedigree is shown in Figure-2 A). Patient II was a five year old girl and she presented in September 2014 with complaints of delay in healing of minor wounds. Patient II developed a habit of biting nails and skin surrounding it (Figure-2 B). Minor skin lesions typically persisted for a long time and developed scabs or infections (Figure-2 C).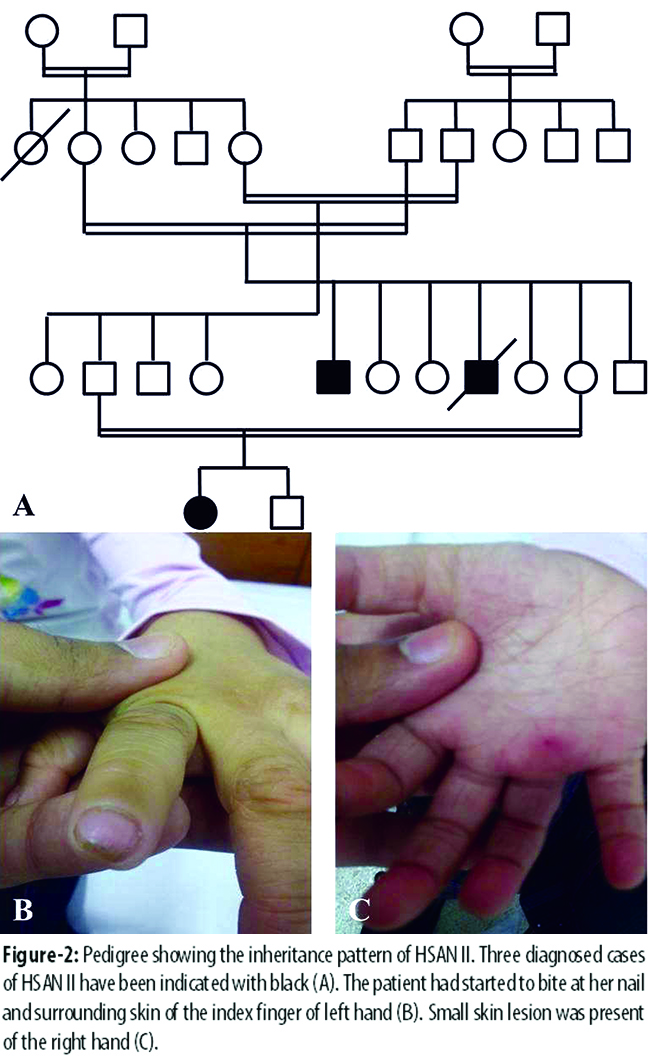
However, the lesions and infections did not progress to the point of shedding of digits or limbs. Clinical examination showed that the patient had complete lack of pain sensation throughout her body along with hyporeflexia; however, no abnormalities in cardiovascular reflexes, pupillary changes, swallowing and casual sweating was observed. Electromyography and nerve conduction studies showed that motor nerve conduction and needle electromyography examination of sampled muscles was normal; however, bilateral sural and radial sensory nerve action potentials were not recordable. Similar to patient I, no other systematic or neurological deficit was reported in individual II. These findings are suggestive of HSAN II.
Discussion
HSAN II is a rare genetic disease with few reported cases worldwide. To the best of our knowledge no cases of HSAN II have been reported from Pakistan. Here we report, two case of HSAN II belonging to one family. The patients identified here have a classical presentation of this disease. Previous studies have shown that the loss of pain, temperature and touch sensation makes HSAN II patients susceptible to injuries and complications that can lead to shedding of digits or even limbs.4 Our patient I had developed similar complications that led to shedding of digits of both his hands and right foot and surgical amputation of left leg. However, patient II showed symptoms of this disease but had not developed any complications.
One weakness of our case report is that we did not conduct any genetic analysis. However, performing a detailed genetic analysis was beyond the scope of the health providers. Although, we do emphasize that genetic studies to identify the mutations associated with this disease are very important. Unlike other subtypes of HSAN, no mutation has been consistently associated with HSAN II. Therefore identification of more cases of HSAN II is crucial in order to discover the definitive mutation responsible for this genetic disorder. Thereafter, in vitro and in vivo studies based on these mutations can help us uncover the mechanisms that lead to this disease and develop a cure for it. We can also use this knowledge induce degeneration of pain receptors or nerve fibers in patients suffering from intractable pain and hence develop a novel pain management strategy.
Conclusion
We have reported two rare cases of HSAN II belonging to a large Pakistani family. Identification of this and other such cases of HSAN II can help us better understand this rare disease and perform further research on its genetics, which can help us to develop treatment options for it. Understanding the mechanisms involved in the degeneration of pain receptors or nerve fibers in HSAN II can also help in developing novel treatment options for patients suffering from intractable pain.
Acknowledgement
We would like to thank Dr. HR Ahmad for his advice and support during the preparation of this case report.
References
1. Rotthier A, Baets J, Timmerman V, Janssens K. Mechanisms of disease in hereditary sensory and autonomic neuropathies. Nat Rev Neurol 2012; 8: 73-85.
2. Rotthier A, Auer-Grumbach M, Janssens K, Baets J, Penno A, Almeida-Souza L, Van Hoof K, et al. Mutations in the SPTLC2 subunit of serine palmitoyltransferase cause hereditary sensory and autonomic neuropathy type I. Am J Hum Genet 2010; 87: 513-22.
3. Axelrod FB, Simson GG. Hereditary sensory and autonomic neuropathies: types II, III, and IV. Orphanet J Rare Dis 2007; 2: 39.
4. Axelrod FB. Hereditary sensory and autonomic neuropathies. Familial dysautonomia and other HSANs. Clin Auton Res 2002; 12: 12-4.
5. Hilz MJ. Assessment and evaluation of hereditary sensory and autonomic neuropathies with autonomic and neurophysiological examinations. Clin Auton Res 2002; 12 Suppl 1: I33-43.
Journal of the Pakistan Medical Association has agreed to receive and publish manuscripts in accordance with the principles of the following committees: