
K.B. Asumal ( Section of Neurology, Department of Medicine, The Aga Khan University Hospital, Karachi. )
M. Wasay ( Section of Neurology, Department of Medicine, The Aga Khan University Hospital, Karachi. )
S.N. Ali ( Section of Neurology, Department of Medicine, The Aga Khan University Hospital, Karachi. )
November 2002, Volume 52, Issue 11
Case Reports
Introduction
Hallervorden Spatz Disease is a rare familial neurodegenerative disorder, which primarily affects children but also can occur in adults1. Major clinical features are abnormal involuntary movements and cognitive impairment2. Diagnosis is based on clinical and radiological features. The radiological features are hypointense signals in globus pallidus and substantia niagra on MR! of brain3. Occasionally the pallidal hypointense signals surround hyperintense signals, this is known as “tiger-eye-sign”4 and is postulated to be specific for Hallervorden Spatz Disease. We report two brothers with such MRI findings.
Case Report
Case 1
A thirteen year, right handed boy presented to the neurology clinic with three year history of progressive walking difficulty and recurrent falls. He also suffered from abnormal involuntary movements, which would disappear during sleep and progressive dysarthria to the extent that his speech was barely intelligible. There was also a history of global cognitive decline. During the six month prior to presentation he became progressively irritable, restless and displayed sudden and inappropriate episodes of laughter. There was no history of fever, jaundice or seizures, His examination was significant for decreased comprehension and attention. His speech was hypophonic and barely intelligible. Generalized dystonia was noted, with ballistic component on right side in association with bilateral blepharospasm. His tone was increased bilaterally although more on the right side. Plantar response was normal. . His fundoscopic examination was normal. His CBC and liver function tests (LFTs) were normal, serum ceruloplasmin and urinary copper levels were also normal (O.243g/L and 23ug/24 hours respectively). He was given a trial of levodopa during 2nd year of his illness without any benefit. An MRI of the brain was performed, which revealed the classic “tiger-eye-sign” in the globus pallidus bilaterally (Figure 1).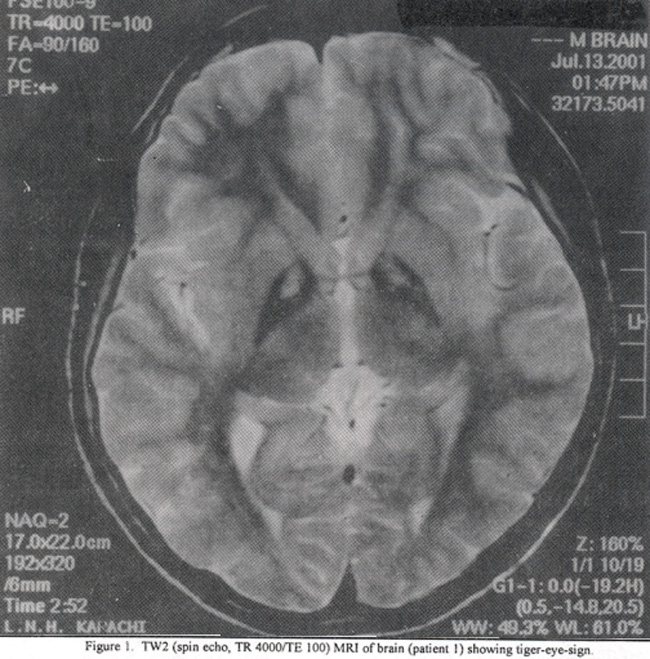
Case 2
An eleven-year boy (younger brother of case 1) presented to neurology clinic with history of progressive walking difficulty, decreased school performance and involuntary movements for about two years, His examination was significant for mild cognitive impairment, generalized dystonia and mild irritability. His fundoscopic examination was also normal. His overall clinical presentation was similar to that of his elder brother (case 1) but was milder in severity. Laboratory workup including CBC, liver enzymes, serum ceruloplasmin and urinary copper levels were normal. His MRI brain showed similar findings i.e., tiger-eye-sign (Figure 2).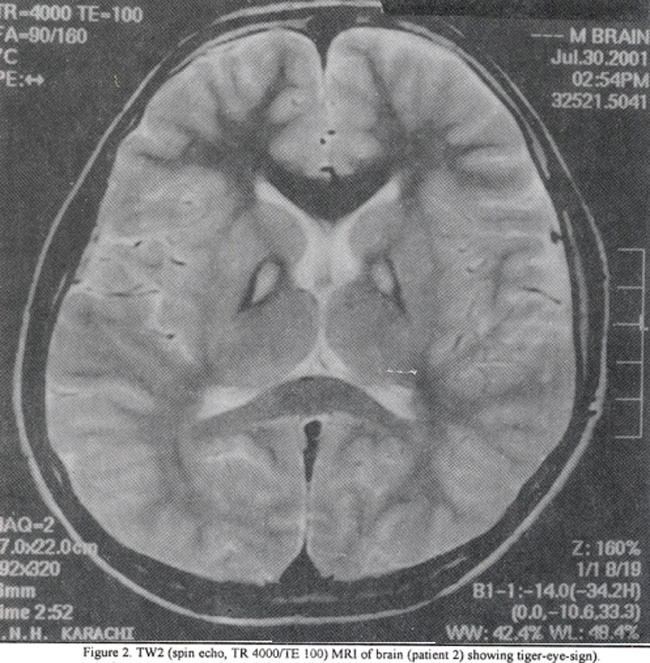
Discussion
Hallervorden Spatz Disease is a rare autosomal recessive disorder, which was first described by Hallervorden and Spatz in a family in 19221. They described it as a familial disorder that began prior to age of 10 years, often associated with clubfoot deformity, gradually increasing stiffness in all limbs, impaired speech and dementia, Its gene is not identified yet though its location is chromosome 205. The most important pathological findings are iron deposition symmetrically in both globi pallidi and substantia nigra (pars reticulata) and distal axonal swelling i.e. spheroids6.
Clinical features include progressive gait differences, rigidity, spasicity, paucity of movement, dystonia, dysarthria, tremor and cognitive impairment2. Retinitis pigmentosa is also present in 20% of patients3. Rarely blephrospasm, ballism, freezing, ptosis and apraxia of eyelid opening are seen3. The clinical presentation mimics several neurological disorders, the most important one being Wilson’s disease. Differentiation between the two disorders is very important, as Wilson’s disease is treatable where as there is no treatment available for Hallervorden Spatz disease. The definitive diagnosis of Hallervorden Spatz disease could only be made on histopathological grounds hence the term Hallervorden Spatz syndrome has been used for the clinical entity, when diagnosis is made on the bases of clinical and laboratory parameters7. The most important investigation, which supports the diagnosis, is magnetic resonance imaging (MRI) of brain. Most common finding on MRI is symmetric hypointense signals in globus pallidi3. Sometimes hyperintense signals lie within these hypointense signals, the so-called “Tiger-eye sign”4,7,8. This sign is said to be specific for the disease. Swiaman devised the diagnostic criteria for the disease based on clinical and radiolological and laboratory findings9. According to the Swiaman criteria,
Hallervorden Spatz disease can be diagnosed if a person meets the obligatory criteria which are:
a) onset during first two decades,
b) progressive course and
c) evidence of extra pyramidal dysfunction,
Along with the obligatory criteria, there must be present at least two of the following eight corroborative criteria:
a) pyramidal tract signs,
b) progressive mental retardation,
c) seizures,
d) retinitis pigmentosa/optic atrophy,
e) positive family history,
f) hypo densities in basal ganglia on MRI brain,
g) abnormal cytoplasm in lymphocytes and
h) sea-blue histocytes on bone marrow
We propose that application of these criteria may result in attributing false diagnosis of an untreatable and rare disease to the patients with treatable and comparatively common diseases like Wilson’s disease and hence these criteria need to be revised. We suggest including the radiological findings as obligatory criterion. The diagnosis of Hallervorden Spatz Disease can be made confidently on the basis of the triad i.e. cognitive impairment, abnormal involuntary movements and the ‘tiger-eye-sign’ on MRI brain.
References
1.Wolfang HO, Molter JC. Other degenerative syndromes that cause parkinsonism. In: Watts RL and Koller WC eds. Movement disorders:
neurologic principle sand practice. New York: McGraw Hilt Publishers, 1997, pp. 331-44.
2.Dooling EC, Schoene WC, Richardson EP. Hallervorden Spatz Syndrome. Arch. Neurol., 1974;30:70-83.
3.Rodintzky RL. Hallervorden Spatz Syndrome. In: Stem MB and Kotter WC eds. Parkinsonian Syndromes. New York: Marcel Dekker, 1993, PP. 341-58.
4.Malandnni A, Fabrizi GM, Salvadori C, et al. Clinicopathotogical study of familial late infantile Hallervorden Spatz disease: a particular form of neuroacanthocytosis. Childs. Nerv. Syst., 1996;12:155-60.
5.Riley DE, Lang AE. Movement Disorders. In: Bradley WG, Daroff RB, Fenichel GM and Marsden CD Eds. Neurology in clinical practice. London: Butterworth-Heinemann,, 2000, pp. 1889-1930.
6.Halliday W. The nosology of the Hallervorden Spatz disease. J. Neurol. Sci., 1995;134:84-91.
7.Sethi KD, Adams RJ, Loring DW, et al. Hallervorden Spatz Syndrome: clinical and magnetic resonance imaging correlation. Ann. Neurol., 1988; 24:692-94.
8.Curatolo P, Feliciani M. Early clinical and imaging (high-field MRI) diagnosis of Hallervorden Spatz disease. Neuroradiology, 1994;36:247-48.
9.Swiaman KF. Hallervorden Spatz syndrome and brain iron metabolism. Arch. Neurol., 1991;48;1285-93.
Related Articles
Journal of the Pakistan Medical Association has agreed to receive and publish manuscripts in accordance with the principles of the following committees: