
Safia Bibi ( PMRC, Research Centre, JPMC, Karachi. )
March 2015, Volume 65, Issue 3
Learning Research
Informed consent is a basic ethical requirement in biomedical research involving human subjects. It drives its value from the basic principle of ethics that human beings are autonomous agents and we ought to respect their autonomy i.e. self-ownership.1 Informed consent in human subject research is therefore designed as a mean to show respect to the autonomy of research participants allowing them to make a free choice regarding participation in research.
It is important to understand that Informed consent is not a mere document that requires signatures from research participants. It is a process that facilitates an individual to make a voluntary informed decision that whether he/she should participate in a research study or not. U.S. Food and Drug Administration (FDA) in its recent revised policy defines the process of informed consent as,
"Informed consent involves providing a potential subject with adequate information to allow for an informed decision about participation in the clinical investigation, facilitating the potential subject\'s comprehension of the information, providing adequate opportunity for the potential subject to ask questions and to consider whether to participate, obtaining the potential subject\'s voluntary agreement to participate, and continuing to provide information as the clinical investigation progresses or as the subject or situation requires."2
The overall process of obtaining informed consent can be divided into three components i.e. competency assessment, disclosure of information and voluntariness.1
Competency Assessment
This step is considered to serve gate keeper\'s role in health research for obtaining a valid informed consent.1 It takes in account the cognitive ability of human subjects and requires that potential research subject must be legally and psychologically capable to understand the proposed study treatment/intervention and make an informed decision after taking in account the risks and benefits associated with the study. In case a person is not competent (e.g. minors, unconscious patients, mentally retarded patients etc.), "Proxy" consent may be obtained from his/her Legally Authorized Representative (LAR) e.g. parents, spouse or next of kin.3 In case of children who are by definition too young to sign an informed consent but are old enough to understand the research and its risks and benefits, their assent in addition to proxy consent from LAR is also required.4 For example if a researcher wants to conduct a study on vitamin D levels of school going children (aged 13-15years), an assent from children in addition to LAR\'s informed consent will be required. Declaration of Helsinki states,
"When a potential research subject who is deemed incapable of giving informed consent is able to give assent to decisions about participation in research, the physician must seek that assent in addition to the consent of the legally authorized representative. The potential subject\'s dissent should be respected."5
Similarly there are certain groups of people who are identified as vulnerable groups e.g. prisoners, refugees, employees etc. who though competent but cannot make voluntary decision based on their position in a particular social set up. It is usually advisable that researchers should not recruit vulnerable groups who carry the risk of duress in a study unless the research question addresses that particular population.6,7
Disclosure
The second step in the process of informed consent is disclosure of study information to the potential subject. This includes all the relevant information that may have an impact on the voluntary decision making of subject regarding the participation in research. According to World Medical Association’s declaration of Helsinki potential subject must be adequately informed of the aims, methods, sources of funding, any possible conflicts of interest, institutional affiliations of the researcher, the anticipated benefits and potential risks of the study and the discomfort it may entail, post-study provisions and any other relevant aspects of the study including the right to refuse to participate in the study or to withdraw consent to participate at any time without reprisal.5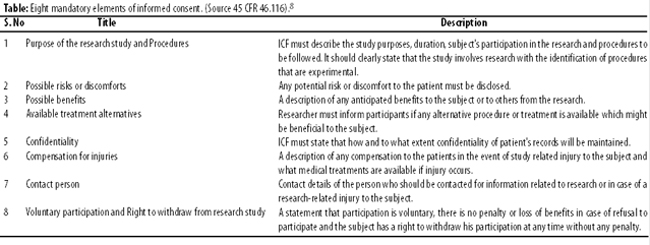
Table describes the eight mandatory elements of an ICF as per FDA\'s 45 CFR 46.116.8
Whether the information provided in an informed consent form (ICF) regarding a particular treatment/intervention is adequate or not can be evaluated on the basis of a reasonable physician standard, a reasonable patient standard and subjective standard. These standards refer to the information that a typical physician, an average patient and that particular patient would want to know if asked to participate in that particular study.9
Example: A researcher studying the prevalence of hepatitis in a particular community later wanted to use the same blood samples to determine the prevalence of AIDS in that community. Institutional Bioethics Committee would not usually allow this as the researcher did not disclose that to the participants while obtaining consent.
In addition to the disclosure of information it is also important to provide the research participant with conducive environment to facilitate voluntary decision making. Dialogue between the person obtaining consent and the potential subject must always be conducted in private, confidential and safe settings.10 Similarly, though it is not necessary that consent be obtained by Principle investigator (PI) but PI must ensure that the person obtaining consent is fully conversant with the study to answer the subject\'s queries. Here it is also important that information be presented in a way that potential subject may understand e.g. simple language be used. Researcher may take the support of videos or flip charts to further simplify or explain procedures. Translating ICF in local language where subject is not able to read or understand the official language is also recommended. Simultaneously comprehension of participant regarding given information must also be ascertained by asking questions.10
Voluntariness: Once a competent subject has been provided with the adequate information about the study then comes the final step of informed consent process i.e. Voluntariness. Voluntariness means a decision free of any type of coercion or undue influence. Ethical research requires that potential subject should voluntarily decide to participate or not to participate in a research without any form of coercion or lucrative incentives. Declaration of Helsinki states,
"After ensuring that the potential subject has understood the information, the physician or another appropriately qualified individual must then seek the potential subject\'s freely-given informed consent, preferably in writing."5
To obtain a coercion free consent it must be ensured that person obtaining informed consent is not in a dependent relationship to the potential subject as there will always be a possibility of some sort of coercion in such a case e.g. physician obtaining consent from his patient, teacher obtaining consent from his student or an employer recruiting his employee for research purpose. In all such cases subject\'s voluntariness may be compromised and he may consent to participate in the research out of the fear of consequences of not consenting.
Similarly undue inducement of research participants through financial incentives or other gains is prohibited. Such inducements compromise voluntariness as potential subjects chose to participate in the study not for the sake of generation of knowledge but due to financial or other gains.11 However, here it is important to make distinction between compensation and inducement. A person may be offered compensation for his participation in research provided it does not have undue influence on potential subject\'s decision regarding the participation in research. For example, provision of health care reimbursement, cost of travel, loss of wages, transfer of any foreseen technology etc do not constitute inducement.
It is also important for a researcher to understand that informed consent is not an isolated event which is completed when participant signs the ICF. The dialogue between the researcher and the subject should continue and subjects must be free to ask questions and seek any further information throughout the study.
Conclusion
The purpose of informed consent in research is to show respect to research participants by allowing them to make free choice but in order to ensure that the purpose is served, it is important for researchers to understand the consenting process. Informed consent is only valid when it is given voluntarily by a competent person after being informed about all the relevant information that could have an impact in shaping his decision regarding the participation in research.
References
1. Beauchamp TL, Childress JF. Principles of Biomedical Ethics (5th ed.). New York, NY: Oxford University Press, 2001.
2. U.S Department of Health and Human Services, U.S. Food and Drug Administration (FDA). Draft Informed consent information sheet, Guidance for IRBs, clinical investigators and Sponsors, July 2014.
3. Glass KC, Ofenberg MS. Incompetent subjects as research subjects and the ethics of minimal risk. Camb Q Healthc Ethics 1996; 5: 362-72.
4. Committee on Bioethics. Informed consent, parental permission and assent in pediatric practice. Pediatrics 1995; 95: 314-7.
5. World Medical Association. Ethical principles for medical Research involving human subjects. (Online) (Cited 2014 Nov 12). Available from URL:http://www.wma.net/en/30publications/10policies/b3/.
6. Schwenzer KJ. Protecting vulnerable subjects in clinical research: Children, Pregnant women, Prisoners and Employees. Respiratory Care 2008; 53: 1342-9.
7. Haque MJ. Research on vulnerable groups: The Medical Researchers View. TAJ 2005; 18: 43-46.
8. U.S Department of Health and Human Services. Basic Health and Human Services (HHS) policy for protection of human research subjects at 45 CFR 46.116. Revised January 15, 2009.
9. De Bord J. Ethics in medicine; Informed consent. University of Washington School of Medicine. (Online) March 2013 (Cited 2014 Nov 12). Available from URL: https://depts.washington.edu/ bioethx/topics/consent.html.
10. Association of Clinical Researsh Professionals (ACRP). The process of informed consent. April, 2013.
11. Draper H, Wilson S, Flanagan S, Ives J. Offering payments, reimbursement and incentives to patients and family doctors to encourage participation in research. Family Practice 2009; 26: 231-8.
Journal of the Pakistan Medical Association has agreed to receive and publish manuscripts in accordance with the principles of the following committees: