
Saqib Hussain Ansari ( Department of Hematology, Thalassaemia Prevention Center, Omair Sana Foundation, Karachi. )
Nida Baig ( Department of Research, Thalassaemia Prevention Center, Omair Sana Foundation, Karachi. )
Tahir Sultan Shamsi ( Department of Hematology, Thalassaemia Prevention Center, Omair Sana Foundation, Karachi. )
Saif-ur-Rehman ( Laboratory and Blood Bank Thalassaemia Prevention Center, Omair Sana Foundation, Karachi. )
Zeeshan Hussain Ansari ( Department of Research, Thalassaemia Prevention Center, Omair Sana Foundation, Karachi. )
Zubaida Behar ( Laboratory and Blood Bank Thalassaemia Prevention Center, Omair Sana Foundation, Karachi. )
Kousar Perveen ( Department of Research, Thalassaemia Prevention Center, Omair Sana Foundation, Karachi. )
Sajida Erum ( Department of Research, Thalassaemia Prevention Center, Omair Sana Foundation, Karachi. )
Zoaib Raza Bukhari ( Laboratory and Blood Bank Thalassaemia Prevention Center, Omair Sana Foundation, Karachi. )
Muhammad Tahir Khan ( Department of Research, Thalassaemia Prevention Center, Omair Sana Foundation, Karachi. )
Mohammad Akbar ( Laboratory and Blood Bank Thalassaemia Prevention Center, Omair Sana Foundation, Karachi. )
December 2012, Volume 62, Issue 12
Original Article
Abstract
Objective: To screen immediate family members of thalassaemia patients for carrier identification and counselling.
Methods: The cross-sectional study was conducted at an urban thalassaemia treatment and prevention centre in Karachi, Pakistan, from January to December 2008, and involved 188 siblings of 100 thalassaemia patients. Complete blood count, including haemogram, was performed in the siblings. Samples with MCV <75fl and MCH <25% were subjected to haemoglobin-electrophoresis. Haemoglobin A2 of 3.5% to 7.0% was labelled as b-thalassaemia minor. Those with haemoglobin A2 of 3.0-3.4% but red blood cell count of >4.5x1012/L were reported as equivocal and were screened for iron deficiency anaemia and a repeat haemoglobin A2 estimation was done on high performance liquid chromatography. Equivocal results of the chromoatography were screened for thalassaemia mutation. Mean values along with standard deviation were worked out for relevant variables.
Result: Of the 188 subjects, there were 124 (66%) males and 64 (34%) females. The mean age was 16.5±6.3 years (range: 3 months to 30 years) and the mean family size was 1.88±3.8 (range: 1-12) children per family. There were 51 (51%) first-cousin marriages in this group. Of the siblings, 65 (34.5%) were identified as normal, while 117 (62.2%) were reported as b-thalassaemia carriers. Six asymptomatic siblings were reported as consistent with b-thalassaemia major.
Conclusion: There were 62.2% siblings identified as beta thalassaemia carriers in the study as opposed to 5-8% carriers in the general population. We also identified six asymptomatic and unidentified cases of b-thalassaemia intermedia in these families. Therefore, in our context where both resources and budgets are limited, it is practical to focus on siblings of identified thalasaemia patients.
Keywords: b-thalassaemia, Carriers, Hb-electrophoresis. (JPMA 62: 1314; 2012).
Introduction
Thalassaemia, the most common hereditary anaemia is characterised by defects in the synthesis of one or more of the globin chains that form haemoglobin (Hb) tetramere. b-thalassaemia, one of the genetic haemoglobinopathies with autosomal recessive pattern of inheritance, is known to be caused by more than 200 point mutations observed at the molecular level.1 Thalassaemia, along with other haemoglobinopathies, is the most common genetic disorder affecting approximately 7% of the world population.2 According to the Thalassaemia International Federation, only about 200,000 patients with thalassaemia major are alive and registered as receiving regular treatment around the world.3 It has a high prevalence in the Mediterranean countries, the Middle East, Central Asia, India, southern China, and the Far East as well as countries along the north coast of Africa and in South America.4 b-thalassaemia is one of the commonest inherited haemoglobin disorder and its statistics in Pakistan present a grim scenario. It has an overall carrier frequency of more than 5%.5 Approximately 40,000 cases of transfusion-dependent children with thalassaemia major are presently registered and each year nearly 5250 are born in the country with a population of about 150 million.6 Homozygote b-thalassaemia patients usually present with severe anaemia within the first year of life. Severe anaemia is incompatible with life; patient requires regular blood transfusion and iron chelating therapy for survival.7 The cost of the treatment is not affordable by an average Pakistani family. This is a major financial burden on the family and society, as treating thalassaemia alone will consume up to about 40% of our current healthcare budget.5 The best treatment option available is bone-marrow transplantation from human leukocyte antigen (HLA)-identical siblings in patients who have no evidence of iron-mediated liver damage. However, this treatment option is beyond the reach of a common person in terms of affordability. The best possible solution is to identify carriers through screening, provide genetic counselling and prenatal diagnosis to reduce the birth rate of affected infants and improve the prognosis of the affected patients.8 Countries like Cyprus, Italy, England, Greece, Iran, Turkey, Sri Lanka, China, Tunisia, Thailand and Malaysia have well-established screening and prenatal diagnostic programmes. With screening and prenatal diagnosis programmes Cyprus, Italy and Greece have been 100% successful and the birth prevalence of homozygote beta thalassaemia now stands at zero.9-11 Worldwide screening and prevention programmes adopt one or more of the following methods: extended and immediate family screening; newborn screening; premarital screening; pre-pregnancy screening; and pregnancy screening (Chorionic Villus Sampling during the first trimester). The aim of all these approaches is to reduce the rate of birth of affected infants. Pakistan does not have a national thalassaemia screening or prevention programme or any policy in this regard. Hence, the present study was designed to identify a cost-effective feasible method for nationwide applicability. We focussed on screening immediate family members of those already affected.
Subjects and Methods
The cross-sectional survey was conducted at the Omair Sana Thalassaemia Treatment and Prevention Centre, Karachi, from January to December 2008. Ethical approval of the study protocol was obtained from the ethical committee of the institution. Since it was a cross-sectional survey, all the enrolled patients\\\' parents were called in for individual counselling. They were provided detailed information about the importance and need of screening and they were also provided information about the laboratory methods and protocol that was to be employed for screening. From among those who agreed to participate and provided informed written consent, 100 families having a child with beta thalassaemia major were randomly selected. On the first meeting with family, the genetic counsellor took family history and filled up the study enrollment form. Each family and subject was given a unique study code. On followup i.e. the second visit, blood samples were collected in ethylenediaminetetraaectic (EDTA)-anticoagulated blood sample tubes. Two families were called in one day.
Screening for thalassaemia was performed using the conventional primary screening method. The blood samples collected were subjected to complete blood count (CBC) and red blood cell (RBC) indices using automatic cell counter Sysmex XF-2100. Samples with mean corpuscular Volume (MCV) < 75fl and mean corpuscular haemoglobin (MCH) <25pg were further studied through Hb-electrophoresis, using cellulose acetate in alkaline buffer. Subjects with Hb A2 of 3.5% to 7.0% were labeled as b-thalassaemia minor or carriers. Those with Hb A2 3%-3.4% and RBC count of >4.5x1012/L were screened for iron deficiency. They received a trial of iron therapy and were checked for Hb A2 both by Hb-electrophoresis and High Performance Liquid Chromatography (HPLC). Equivocal results on HPLC were further screened for thalassaemia mutations by Multiplex Amplification Refractory Mutation System (ARMS).
Results
Out of the 188 siblings of homozygous b-thalassemic index cases, there were 124 (66%) males and 64 (34%) females. The mean age of the study group was 16.5±6.3 (range: 3 months and 30 years). The mean family size was 1.88±3.8 (ranging between 2 and 12) children per family. There were 51 (51%) first Cousin marriages in the 100 families. Among the siblings, 65 (34.5%) were identified as normal, whereas 117 (62.2%) were reported as b-thalassemic carriers (Table-1).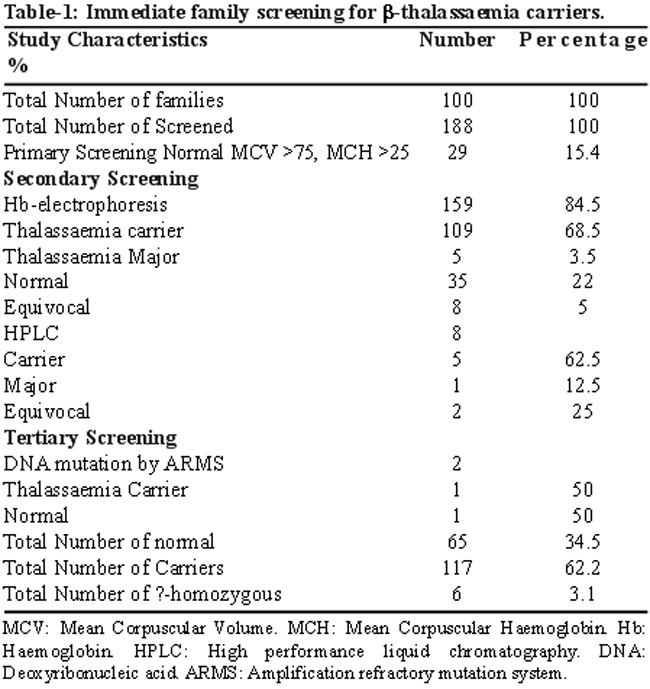
The mean Hb, MCV, MCH and Hb A2 values in the carriers were 10.7 ± 1.4, 66 ± 8, 19 ± 2.5 and 11.9 ± 0.5 respectively whereas in normal individuals they were 11.8 ± 1.7, 79 ± 9, 25 ± 3.5 and, 2 ± 0.01 respectively (Table-2).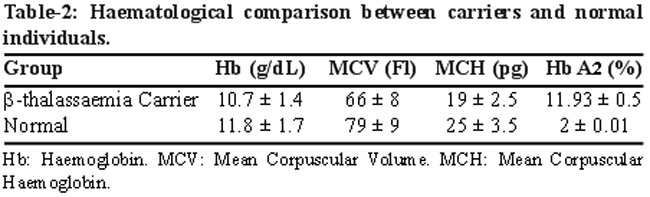
Six asymptomatic siblings were identified as being consistent with b-thalassaemia major/intermedia (Table-3).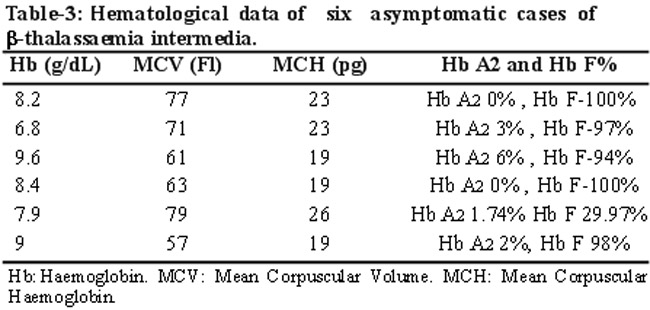
Discussion
Preventive screening programmes to identify carriers are being used by many countries where thalassaemia is a common disease. The various approaches used are population screening, high-risk group screening, antenatal screening, and extended family screening (cascade screening). For carrier identification, studies have used extended family screening (inductive screening) in a Pakistani population with high rate of consanguineous marriages.5-12 They were successful in identifying 31% and 44.4% of thalassaemia carriers respectively among the relatives of thalassaemia children. An Indian study carried out pre-marital screening for thalassaemia carriers in extended family members of previously diagnosed cases, unmarried adult cases of anaemia attending the hospitals\\\' outpatient department (OPD) and adult college students.13 The yield of carriers from extended family, OPD and college students was 58.6%, 16.1% and 2.87% respectively. Through extended family screening, another Indian study screened 691 individuals and identified 151 (21.9%) as being carriers.14
One study in India studied attitudes of 200 families of index cases towards extended family screening;15 20 percent expressed an unfavourable reaction and preferred to be secretive about their child having thalassaemia to avoid social stigmatisation. Similar responses were witnessed by us; the main reason being the lack of awareness and education among parents and relatives of children with thalassaemia major.16 Immediate family screening is a way forward as evidenced by identification of 62.2% of siblings being carriers as opposed to 5-8% carriers in the general population. On the basis of the prevalence of b-thalassaemia carriers reported earlier,5 we would have had to screen about 1880 people in the general population to identify 100 carriers. Further, minimal efforts were required to create awareness and to do counselling in our group of individuals.
This is not to advocate the abandonment of extended family screening altogether; rather, it\\\'s a strategy to overcome this roadblock as 96% parents of affected children after being counselled and educated about the disease were ready to share information with their relatives.17 School screening programmes for carrier identification in France18 and Canada19 have accomplished success, whereas the Indian experience did not have the desired impact.20 Therefore, this seems to be one of the most cost-effective and practical approaches to identify beta thalassaemia carriers and to start a national screening programme. Evidence exists in the literature for acceptance of screening programmes, prenatal diagnosis and genetic counselling by the immediate and extended family of the index cases. The need of the hour is to use public-private partnership to develop a good network for the optimum care of children with b-thalassaemia, for education, for screening to identify carriers and for genetic counselling in a cost-effective way.
Other options for screening need to be considered and their feasibility needs exploration. Those include targeting mainly school and college students, screening of pregnant women in antenatal clinics, and screening carriers in early pregnancy to avoid women being stigmatised or having to conceal their carrier status.21,22 Only then, we can have a well coordinated national approach towards screening which can be integrated into health services for patients just like immunization plans. Thalassaemia screening can even be made mandatory as well. An important component of a screening programme is to identify and counsel carriers using combination of approaches. Special emphasis needs to be laid on public education and public health surveillance. Community education needs to be clear and precise for the public to completely understand and comprehend the take-home message along with the aims and objectives of the programme. Lessons should be learnt from the failure of insufficiently planned and poorly communicated Sickle Cell Screening campaign of USA back in 1970. The difference between carrier status and homozygous condition needs to be clear to the lay population to avoid carriers being stigmatised and screening being rejected by the target population.23
Conclusion
Screening of immediate family members of thalasaemia patients is more effective than screening in other groups, and, hence, has the potential to lead to a nationwide programme for control of thalassaemia and related haemoglobinopathies. For the successful implementation of any prevention and screening programme, services should not vary in approach or executed in a piecemeal fashion.
References
1. Higgs DR, Thein SL, Woods WG. The molecular pathology of the thalassaemias. In: Weatherall DJ, Clegg B, eds. The thalassaemia syndromes. 4th ed. Oxford, England: Blackwell Science, 2001: 133-91.
2. Weatherall DJ, Clegg JB. 2001. Inherited hemoglobin disorders: an increasing global health problem. Bull World Health Organ 2001; 79: 704-12.
3. Thalassaemia International Federation: Guidelines for the clinical management of thalassaemia, 2nd ed. 2008. (Online) 2008 (Cited 2011 January 3). Available from URL: http:// www.thalassaemia.org.cy.
4. Renzo G, Origa R. Beta-thalassaemia. Orphanet J Rare Dis 2010, 5: 11.
5. Ahmed S, Saleem M, Modell B, Petrou M. Screening extended families for genetic hemoglobin disorders in Pakistan. N Eng J Med 2002; 347: 1162-8.
6. Alwan A, Modell B. Community control of genetic and congenital disorders. EMRO technical publication series 24 Alexandria, Egypt: WHO Regional Office for East Mediterranean, 1997.
7. Olivieri NF. The beta-thalassaemia. N Eng J Med 1999; 341: 99-109.
8. Cao A, Rosatelli MC, Galanello R. Control of beta-thalassaemia by carrier screening, genetic counselling and prenatal diagnosis: the Sardinian experience. Ciba Found Symp 1976; 197: 137-55.
9. Perera DM, Gunasekara D, Wijekoon A, Petrou M. Thalassaemia control in developing countries - the Sri Lankan perspective. Ceylon Med J 2000; 45: 12-6.
10. Vaz FE, Thakur CB, Banerjee MK, Gangal SG. Distribution of beta thalassaemia mutations in the Indian population referred to a diagnostic center. Hemoglobin 2000; 24: 181-94.
11. Weatherall DJ. Keynote address: The challenge of thalassaemia for developing countries. Ann N Y Acad Sci 2005; 1054: 11-7.
12. Baig SM, Din MA, Hassan H, Azhar A, Baig JM, Aslam M, et al. Prevention of beta-thalassaemia in a large Pakistani family through cascade testing. Community Genet 2008; 11: 68-70.
13. Tamhankar PM. Agarwal S, Arya V, Kumar R, Gupta UR, Agarwal SS. Prevention of homozygous beta thalassaemia by premarital screening and prenatal diagnosis in India. Prenat Diag 2009; 29: 83-8.
14. Gorakshakar C Ajit, Colah B Roshan. Cascade screening for ?-thalassaemia. A practical approach for Identifying and counseling carriers in India. Ind Jou of Comm Med 2009; 4: 354-6.
15. Sangani B, Sukumaran Pk, Mahadik C, Vas F, Modell B, Merchant SM, et al. Thalassaemia in Bombay: the role of medical genetics in developing countries. Bull World Health Organ 1990; 68: 75-81.
16. Arif F, Fayyaz J, Hamid A. Awareness among parents of children with thalassaemia major. J Pak Med Assoc 2008; 58: 621-4.
17. Saxena A, Phadke SR. Feasibility of thalassaemia control by extended family screening in Indian context. J Health Popul Nutr2002; 20: 31-5.
18. Lena-Russo D, Badens C, Aubinaud M, Merono F, Mattei JF, Martini N, et al. Outcome of a school screening programme for carriers of haemoglobin disease. J Med Screen 2002; 9: 67-9.
19. McCabe L. Efficacy of a targeted genetic screening programme for adolescents. Commet on [Twenty year outcome analysis of genetic screening programmes for Tay-Sachs and beta-thalassaemia disease carriers in high schools]. Am J Hum Genet 1996; 59: 762-3.
20. Colah R Thomas M, Mayekar P. Assessing the impact of screening and counselling high school children for beta-thalassaemia in India. J Med Screen 2007; 14: 158.
21. Modell B, Harris R, Lane B, Khan M, Darlison M, Old J, et al. Informed choice in genetic screening for thalassaemia during pregnancy: audit from a national confidential inquiry. BMJ 2000; 320: 325-90.
22. A Murtz J. Matching scheme solves Tay Sachs problems. JAMA 1987; 258: 2636-7.
23. Atkin K, Ahmad WI. Genetic screening and hemoglobinopathies: ethics, politics and practice. Soc Sci Med 1998; 46: 445-58.
Journal of the Pakistan Medical Association has agreed to receive and publish manuscripts in accordance with the principles of the following committees: